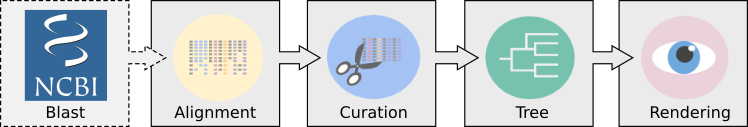
NGPhylogeny.fr is a webservice dedicated to phylogenetic analysis. It provides a complete set
of phylogenetic tools and workflows adapted to various contexts and various levels of user
expertise. It is built around the main steps of most phylogenetic analyses:
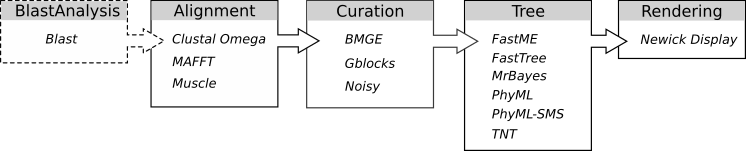
NGPhylogeny.fr integrates several tools for each steps of the workflow:
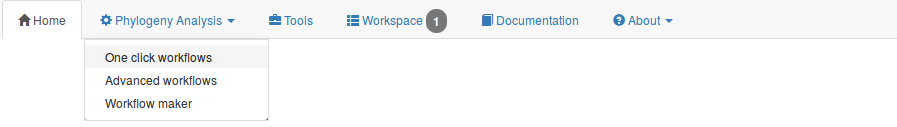
Different ways of using NGPhylogeny.fr are offered, depending on the user needs or expertise:
Oneclick workflows are already preconfigured with default options that should work on the majority of usecases. The only required
input is the sequence data file in Fasta format. Input data type (dna or protein) is detected automatically;
Advanced workflows have basically the same structure as oneclick workflows, but can be parametrized.
It means that the user should customize the options of each step of the workflows: alignment, curation, tree inference.
Workflow maker allows the user to choose the combination of tools that suits best his/her needs, and to customize the parameters.
Moreover, NGPhylogeny.fr provides a user-friendly visualization layer specific to the different
kinds of data usually manipulated in phylogenetics (i.e. alignments, trees).
Finally, Blast-Search module is placed upstream phylogenetic workflows and aims at searching for sequences that are similar to a given user input sequence. Blast-Search then analyses Blast results and builds a quick (and inaccurate) tree in which users can remove unwanted sequences. Remaining sequences may then be used as input of any ngphylogeny.Fr workflows.
Branch supports
In addition to their respective bootstraps, almost all tree inference tools are proposed with the following branch support computations:
For example, it is possible to compute FBP and TBE supports with FastTree.
Bootstrap options are accessible via "Advanced workflows" and "Workflow maker".
Computations
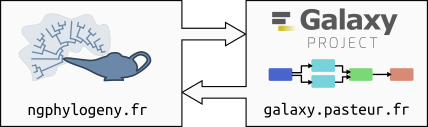
NGPhylogeny.fr works together with Institut Pasteur Galaxy instance to:
Manage tools and workflows;
Run tools and workflows on the underlying computing cluster;
Keep track of run histories.
Oneclick workflows
One click workflows are accessible via the "Phylogeny Analysis/One click workflow" link on the tool bar:
The 4 oneclick workflows implemented in NGPhylogeny.fr differ by the tree inference tool:
PhyML+SMS: This workflow uses PhyML+SMS to select the best evolutionary model and to infer the trees. However, it may not handle very large datasets, as the tree inference may take a very long time. SH-like aLRT branch supports are computed by default;
PhyML: This workflow uses PhyML to infer trees. Default options depend on data type (dna, protein). Like PhyML+SMS, large datasets may not be analyzed with this workflow;
FastME: This workflow infer trees using FastME. FastME provides distance algorithms to infer phylogenies and can work with large datasets;
FastTree: This workflow runs FastTree to infer trees. "FastTree infers approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences" and works for very large datasets. Local branch supports (SH) are computed by default by FastTree.
Sections below describe these oneclick workflows and all the steps.
PhyML+SMS
Workflow outputs:
MAFFT:
Alignment (FASTA)
Guide Tree (TXT)
Output logs (TXT
BMGE:
Cleaned sequences Html (HTML)
Cleaned sequences Nexus (NEXUS)
Cleaned sequences Fasta (FASTA)
Cleaned sequences Phylip (PHYLIP)
PhyML+SMS:
Output logs (TXT)
Output tree (NEWICK)
SMS model comparison (TXT)
SMS best model (TXT)
Newick Display
Tree image (SVG)
PhyML
Workflow outputs:
MAFFT:
Alignment (FASTA)
Guide Tree (TXT)
Output logs (TXT
BMGE:
Cleaned sequences Html (HTML)
Cleaned sequences Nexus (NEXUS)
Cleaned sequences Fasta (FASTA)
Cleaned sequences Phylip (PHYLIP)
PhyML:
Output logs (TXT)
PhyML statistics (TXT)
Output tree (NEWICK)
Newick Display
Tree image (SVG)
FastME
Workflow outputs:
MAFFT:
Alignment (FASTA)
Guide Tree (TXT)
Output logs (TXT
BMGE:
Cleaned sequences Html (HTML)
Cleaned sequences Nexus (NEXUS)
Cleaned sequences Fasta (FASTA)
Cleaned sequences Phylip (PHYLIP)
FastME:
Output logs (TXT)
Distance Matrix (TXT)
Output tree (NEWICK)
Newick Display
Tree image (SVG)
FastTree
Workflow outputs:
MAFFT:
Alignment (FASTA)
Guide Tree (TXT)
Output logs (TXT
BMGE:
Cleaned sequences Html (HTML)
Cleaned sequences Nexus (NEXUS)
Cleaned sequences Fasta (FASTA)
Cleaned sequences Phylip (PHYLIP)
FastTree:
Output logs (TXT)
Output tree (NEWICK)
Newick Display
Tree image (SVG)
Default options
MAFFT
MAFFT flavour: auto
Gap extension penalty: 0.123
Gap opening penalty: 1.53
Direction of nucleotide sequences : do not adjust direction
Matrix selection : No matrix
Reorder output: No
Output format: FASTA
BMGE
Sliding window size: 3
Maximum entropy threshold: 0.5
Gap rate cutoff: 0.5
Minimum block size: 5
If sequence type is DNA:
Matrix: PAM250
If sequence type is Protein or Codon:
Matrix: BLOSUM62
PhyML+SMS
Statistical criterion to select the model : AIC
Tree topology search : SPR
Branch support: No branch support
PhyML
Proportion of invariant sites : Estimated
Number of categories for the discrete gamma model : 4
Parameters of the gamma model: estimated
Tree topology search: SPR
Optimise parameters: tlr
Statistical test for branch support: No branch support
If Nucleotide sequence:
Transition/transversion ratio: estimated
Substitution model: GTR
Equilibrium frequencies: Empirical
If amino-acid sequence:
Model: LG
Equilibrium frequencies : ML model
FastME
Model :
if dna: TN93,
if protein: LG
Equilibrium frequencies: estimated
Gamma distributed rates across sites: Yes
Gamma distribution parameter: 1.0
Remove gap strategy: Pairwise deletion of gaps
Starting tree: BioNJ
Tree Refinement: BalME SPR
Bootstrap: No
Decimal precision for branch lengths: 6
FastTree
Model: if nucleotide sequence: gtr, if amino-acid sequence: lg
Use Gamma distribution: Yes
Branch support: No Branch Support
Advanced workflows
Advanced workflows are accessible via the "Phylogeny Analysis/Advanced workflows" link on the tool bar:
Advanced workflows have the same structure as oneclick workflows, but some options can be customized.
In particular, it is possible to perform specific bootstrap analyses:
Specific branch supports depending on the tree inference program:
aLRT
SH-like
aBayes
Workflow Maker
The workflow maker is accessible via the "Phylogeny Analysis/Workflow maker" link on the tool bar:
In this mode, users can choose the tool they want to run at each step of the workflow:
Multiple alignment: Clustal omega; MAFFT; MUSCLE;
Alignment curation: BMGE; Gblocks; Noisy;
Tree inference: FastME; FastTree; MrBayes; PhyML+SMS; PhyML; TNT;
Tree rendering: Newick Display.
In addition, parameters of each step should be specified.
Launch individual tools
Tools that make up the workflows can be configured and run independently. They are available via the "tools" link on the toolbar:
Blast-Search
Blast-Search is accessible via the "Phylogeny Analysis/Blast" link on the tool bar:
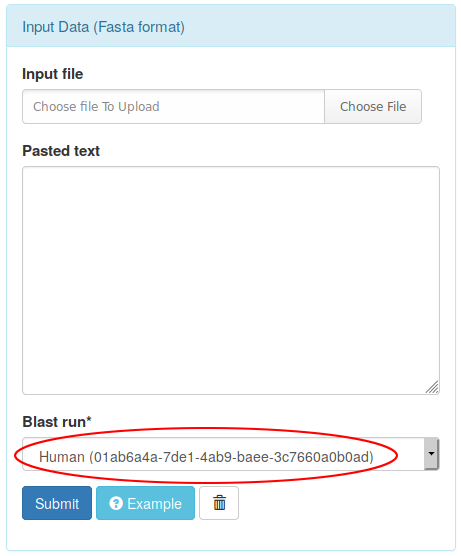
User just have to paste an input sequence, and specify a few options:
Program: blastp, blastn, blastx, tblastn;
Database: refseq, swissprot, nr, etc.;
Max number of results: Only the specified number of closest sequences will be taken into account (based on bitscore);
E.Value threshold;
Query coverage threshold: percentage of aligned sequence compared to query length;
Contact Email: Optional parameter, to be notified when analysis is over.
A Blast run will be launched on the Galaxy instance. The given number of best matches will be treated, and a neighbor joining tree will be computed from a distance matrix (Kimura distance for DNA sequences and Jukes Cantor distance for Protein sequences).
Once the neighbor joining tree is computed, it is displayed in a dynamic visualizer in which clades to remove can be selected individually. Clicking the "Delete selected sequences" button with then remove the sequences from the dataset.
This sequences can be used as input of any phylogenetic workflows by selecting the given blast analysis in the input panel, and then submiting the job:
Typical analysis
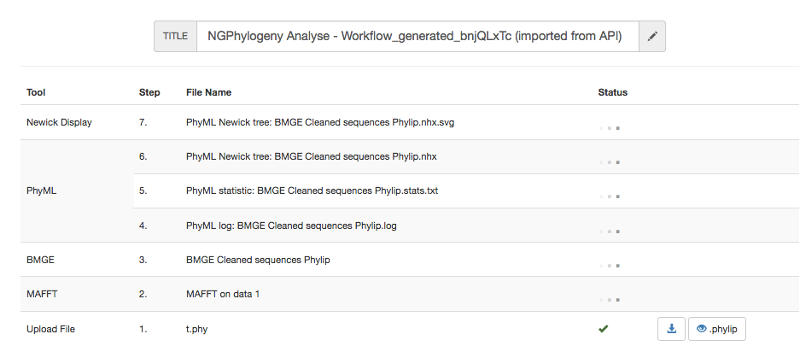
Once the phylogenetic workflow is configured and launched, user is redirected to a waiting page giving informations about the run:
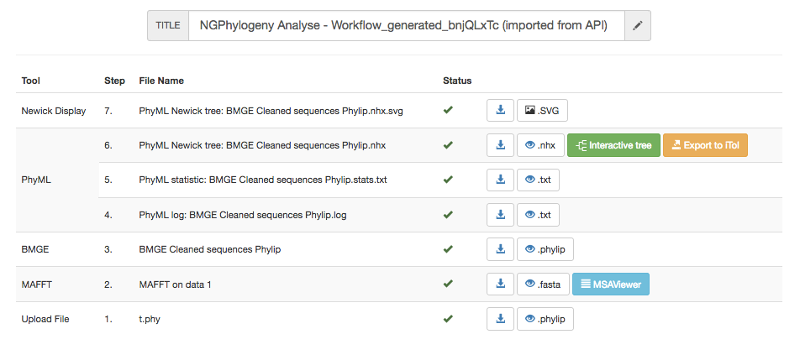
All workflows start by uploading input data to Institut Pasteur Galaxy server. Each step of the workflow is then put in pending status, waiting for available resources on the Galaxy server.
Once a step executed, corresponding result files are downloadable or viewable depending on the format. Images, trees, and alignments are viewable through specific viewers. In addition, trees may be uploaded to iTOL for further investigations.
Run NGPhylogeny.fr locally (Docker)
It is possible to run NGPhylogeny.fr via two docker images:
NGPhylogeny can handle large datasets. However, on the public server, we implemented limitations on the number and length of sequences that depend on the running tool. An error is displayed if the dataset is too large to be analyzed with the public instance of NGPhylogeny.fr.
In contrast, these limitations are not activated if you run NGPhylogeny.fr locally using Docker.